A Million Mutants in a Milliliter
Imagine if you could mutate every single base in a genome and measure how each mutation affects the growth and function of the organism in a quick and easy way. I will discuss a recent work that did exactly this!
Saturating mutagenesis, which sounds like a great band name, is a process in which every possible single mutation is generated in a population of a species. Saturated mutagenic libraries can help researchers identify key mutations that provide growth advantages in certain environmental conditions, such as in the presence of antibiotics, and can help decode gene function in the cell. This has historically been very difficult to achieve, with people using CRISPR libraries and extremely large population sizes to achieve only a partial library of mutants. CRISPR, the premier genome editing technology, makes double-stranded break at specific locations near a PAM, and then a supplied template can edit the mutation into the cut site. CRISPR can only edit near a PAM site, which even for short PAMs limits genomic access, and also has limited efficiency due to cell toxicity and gRNA binding. PAMless CRISPR strategies exist but aren’t mature. Therefore people have used large-volume random mutagenesis, but the working volume required to encapsulate the library is limiting, requiring 8L to work with reliably. The authors of this paper came up with a clever way to fit an entire saturating mutagenesis library into only 1 mL of inoculum, which I will discuss in this post.
The authors of this work are from Pioneer Labs, a focused research organization (FRO) focused on making microbes to both terraform Mars and process Martian soil into usable materials. To do this, they need to first design a microbe that can withstand the harsh Martian environment. Until AI models can generate Mars resistant genomes de novo, they are stuck using evolution. They have made a Mars medium and use adaptive laboratory evolution (ALE) to evolve E. coli and other bacterial species to survive and thrive in Martian-like conditions. However, these methods are not perfect. ALE often optimizes for a certain environment but introduces unnecessary mutations in the process, making the microbe less efficient. Reproducing evolved phenotypes by reintroducing all candidate mutations is laborious, especially when so many may be benign. They were looking for a way to explore more of the fitness landscape all at once while attributing phenotypic changes to specific mutations.
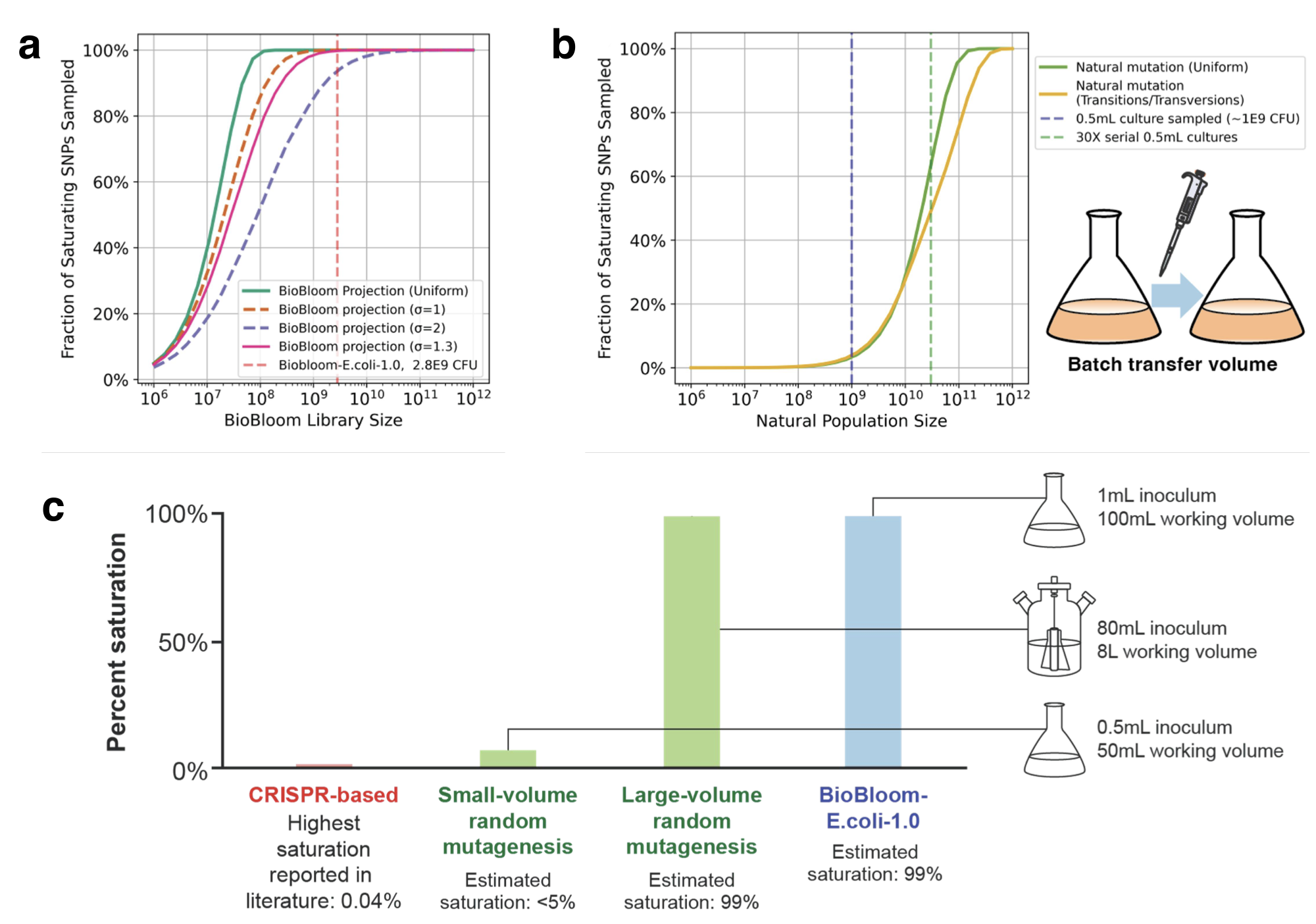
They wanted to explore as much of the fitness landscape as possible, and therefore fit as many mutation combinations into a single library as possible. Let’s do some math to see what’s feasible. To construct a complete library of every single mutation in the genome, each base in the genome needs to be varied to 3 other bases. With E. coli having a genome of size $4.6 \times 10^6$ bp, you need to generate $1.4 \times 10^7$ SNPs. If each cell were a mutant harboring a unique SNP, that means we would need $1.4 \times 10^7$ in our library, although more cells are desired for redundancy (editing efficiency, unequal growth, etc…). Luckily there are about $10^9$ cells per mL of saturated E. coli culture in LB, which would easily fit the library many times over. If we wanted to explore more of the fitness landscape and construct a library of every combination 2 mutations in the genome, our library balloons to $9.8 \times 10^{13}$ cells, and would take 98L of saturated E. coli culture to encapsulate the library, which is far too large. So they cooked up a way to put every single mutation possible in the genome into a single library, since it was theoretically feasible in a small volume.
What came out of the oven was BioBloom, a retron recombineering method that introduces every possible point mutation at every possible place in the genome. By saturating an organism with single mutation mutants, the researchers can identify the fitness effect of each possible mutation in the entire genome. To do this, they used reverse transcriptase to make single stranded DNA from a region of an expressed RNA template (pro-donor). The template is a library of sequences that contain every 82 bp “window” of the E. coli genome with a degenerate “N” as its central base. The ssDNA then complexes with single stranded annealing proteins, and then binds to replicating genomic DNA to create precise edits. The cell retains the plasmid for selection purposes, but it also serves as a barcode since the pro-donor sequence can identify the cell’s SNP.
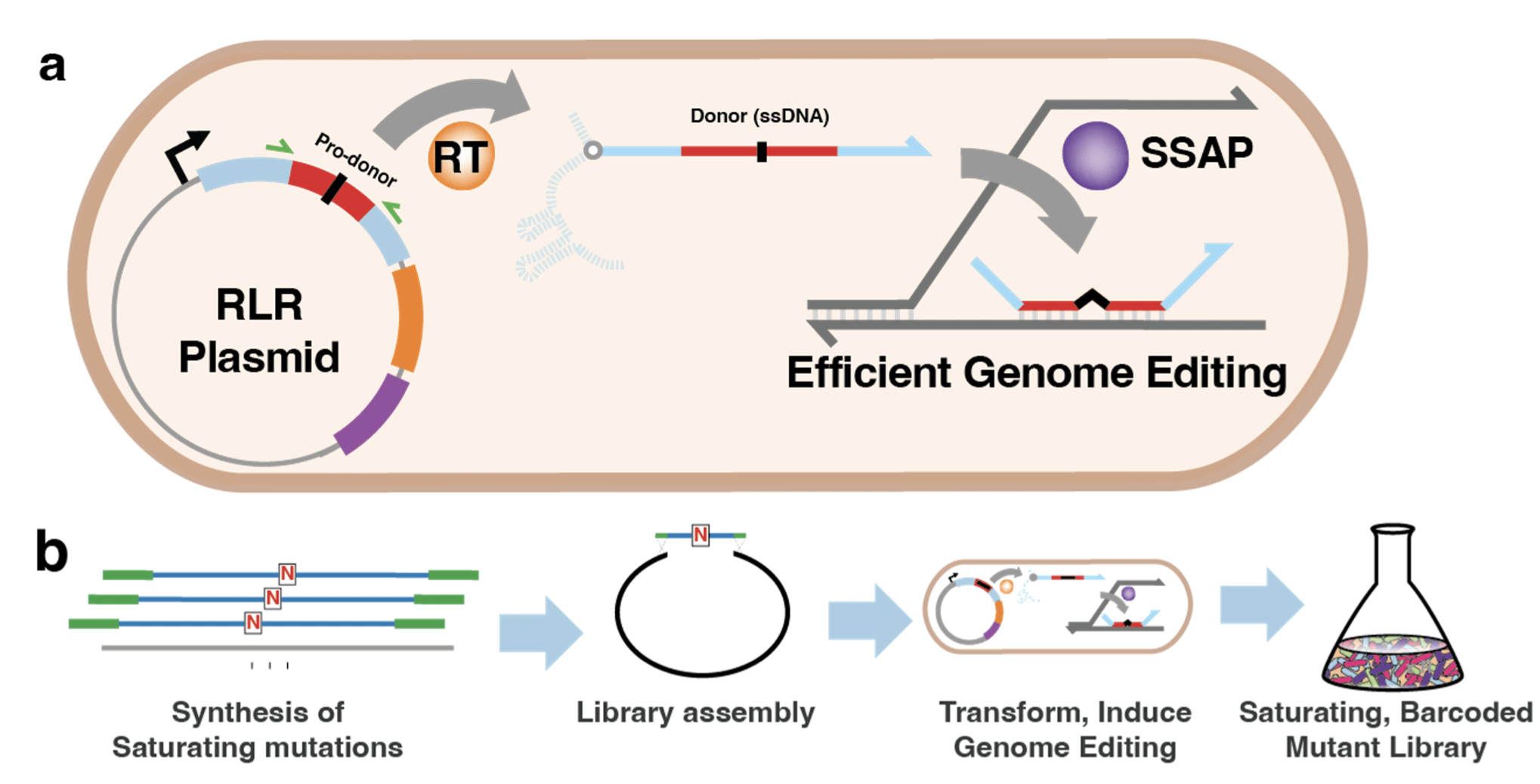
Editing isn’t 100% efficient: other studies using retron recombineering reported 50% editing efficiency, so they used this as a working estimate for their library. If edits are 50% efficient, then $>99\%$ of SNP mutants can be captured in 1 mL of saturated culture. They note that even if the editing efficiency was 10%, $>90\%$ of SNPs can still be captured in a 1 mL saturated culture. This is a large difference between current methods of saturating mutagenesis (random mutagenesis), which require 82mL of saturated culture to capture a similar portion of the library. In addition, the plasmid pro-donor sequence acts as a barcode, which allows them to use simple amplicon sequencing to quantify mutation frequency as opposed to WGS or similar for ALE mutants.
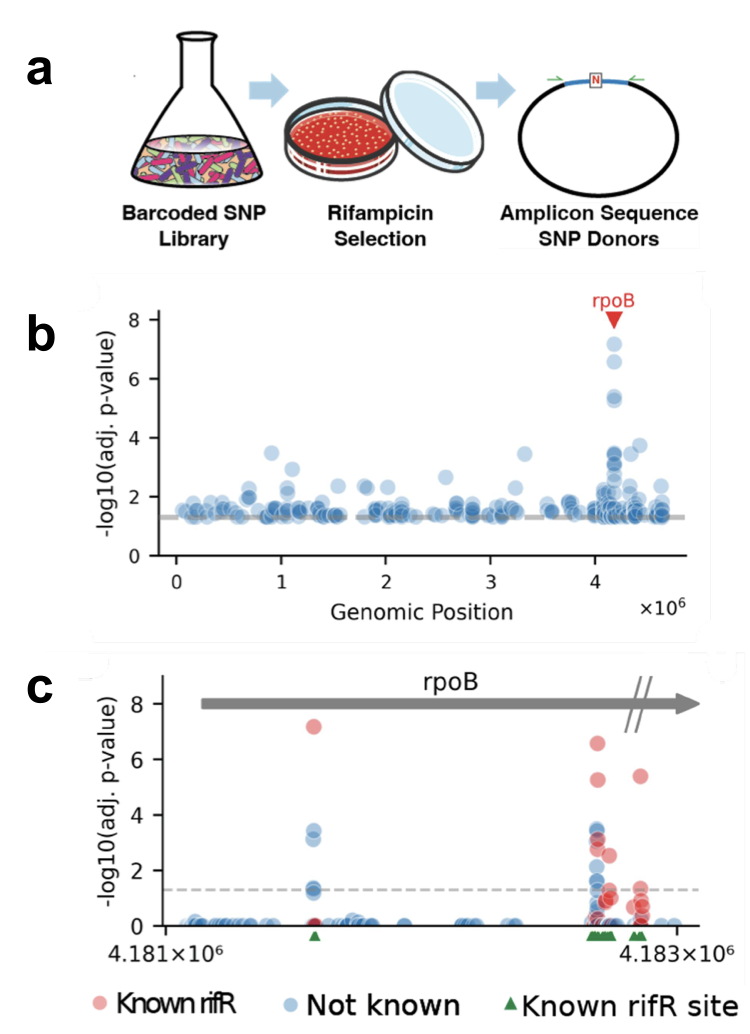
To validate their method, they aimed to rediscover rifampicin resistance mutations, which are well characterized as rifampicin is a first-line tuberculosis (TB) antibiotic. Resistance to rifampicin is much harder to treat and has a higher mortality rate than drug-susceptible TB. In their experiments, they primarily looked in the rpoB locus, which accounts for the majority of known high-resistance mutations. When they selected their library on rifampicin, they sequenced the surviving mutants and found 38 SNPs in the rpoB locus, of which half are known rifampicin resistant alleles whereas the other half have never been reported as resistance conferring mutations. The 19 known SNPs that they rediscovered represents $\sim 25\%$ of known rifampicin resistant mutations, revealed in a single experiment! However it is noted that there are actually 69 known SNPs that confer rifampicin resistance in the rpoB locus. This could be attributed to inefficient editing producing a gap in the library. When they compared these results to a small batch of mutagenized culture, mutagenesis had only produced 4 rifampicin resistant SNPs, far inferior to the BioBloom method (by $\sim$10x).
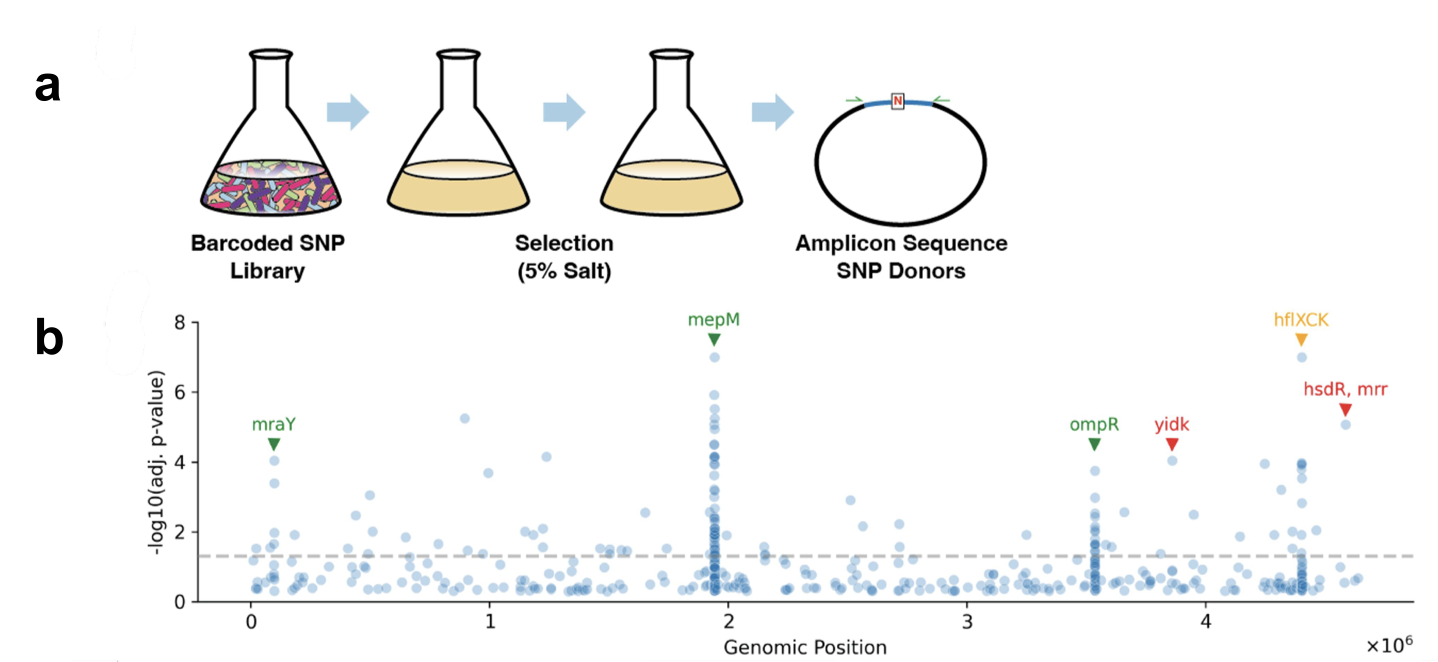
After validation, they wanted to apply the method to a more open ended problem that was expected to surface a variety of mutations, so they grew BioBloom in a salty environment to find salt tolerant mutations. They found 129 SNPs in multiple loci across the genome, some in genes that have been identified in the literature to be involved in salt tolerance and some that haven’t been previously implicated. They took 8 high significance mutants that are suspected to be true hits (in relevant loci) and 2 high significance mutants that are suspected to be artifacts (not relevant loci, no other high significance mutants in the same loci). By growing these mutants in increasingly salty conditions, they found that the half-maximal inhibitory concentration ($IC_{50}$) increased by about $85\%$ in some mutants, even some mutants whose loci had never been connected to salt tolerance before! In others, the increase was more modest. In the suspected artifacts, there was no significant improvements to salt toleration in their assay, confirming their artifact-ness. The authors say that these mutants being high significance may have been due to background mutations arising during editing or strong skew in the starting library. Curiously, the successful mutants didn’t display a greater growth rate or decreased lag time compared to WT, but simply grew to a higher terminal density. Comparing these results to a 1 month long evolution experiment of E. coli in 5% salt media, they found overlapping mutated loci and similar $IC_{50}$ results. The comparison isn’t exactly $1:1$ due to the difference in parental strain salt tolerances. Regardless, they were able to achieve similar results, attribute the phenotypes to singular SNPs, and find the same SNPs more quickly. I wonder if there would be synergistic effects from combining these successful SNPs found in the experiments.
With their method validated and used to find new salt-tolerant loci, they convinced me that this is a powerful method that can be applied in many different ways! Although I wish they had done some more analysis on their initial library so they could estimate editing efficiency with more than just comparisons to other studies and compared pro-donor amplicon abundances from before and after the experiment to quantify mutant frequency in all samples. Although, even with pro-donor frequency quantified, there is no guarantee that the edit actually occurred during the induction period until further sequencing is done. After the study, they improved the method slightly by spreading skew out between replicates and deep sequencing pro-donors to obtain mutant frequency in the initial library. They then deposited this improved library to Addgene for anyone to order.
Overall, this is a really cool method with a lot of different applications. This seems like a great way to initially find which loci are involved in a particular process, and then use that information as a basis of future evolution experiments, using OrthoRep or similar tool for directed mutagenesis. Since this also is a way to quickly find which loci are responsible for which phenotypes, it could also be used to annotate gene function, protein domains, regulatory elements, and probe pathways.
It seems like the main gating cost to making this library again or in other organisms is the cost of the primers that mutate each base with each possible other base to make the RLR plasmids ($\sim \$30\text{k}$!). Since they already have the primers, I wonder if they can explore the combinatorial space of mutations in successive library generation. For example, they would find certain single mutations that allow an organism to grow faster/higher density in a certain environment. They then clone those single mutations into an organism with no background mutations and use the same plasmids to make the library again. This way, they can find which other mutations can work synergistically with the ones they have already found. They can then go further down an evolutionary path until a maxima is reached. This is a more practical way of searching a larger combinatorial space since, as explained above, even every combination of 2 base mutations in the genome becomes unfeasibly large to probe.
I wonder how hard it would be to put this into organisms other than E. coli. The main gating factor would be the $\$30\text{k}$ price tag, but also if the RT and SSAP machinery works in diverse organisms. If there was a substantial effort to collect this SNP data from lots of different organisms, an AI model could be trained on the growth data and predict responses to perturbations or even suggest SNPs towards a certain end goal i.e., growth on Mars.
I cannot help but let my imagination get the best of me and wonder if there was a way to make a library depth of all possible bases in every combination, to probe the most optimal 4 Mb organism possible. What might this organism look like? How might it behave? What unique functions and mechanisms might it have? Unfortunately, it is far too large a number to probe. For any curious reader, this would be $4^{4,000,000}$, which is far too large to write out as it possesses 2.4 million digits.
I am excited to see how the scientific community uses this library for future studies. Can’t wait to see what the folks at Pioneer Labs cook up next to advance humanity to be a multi-planetary species!